Non-Invasive Prenatal Testing (NIPT) is an alternative to amniocentesis, allowing for genotyping of a fetus using a plasma sample from the pregnant mother1. The results of genotyping this sample is an amalgamation of reads from both the fetus and the mother. The mother's DNA predominates, making up approximately 70 - 95% of the available reads. The other reads are the fetal fraction, but half of those are from the mother and cannot be identified as fetal. This leaves approximately 2 - 15% of the DNA as an identifiable fetal fraction (allelic sequence reads present in the plasma and absent in the mother.)
Enhancing the fetal fraction makes fetal genotyping easier, and can be done by using In-silico Size Selection (ISS). The premise is that on average the fetal reads are slightly shorter than maternal reads2. By selecting only the shorter reads the percentage of reads that are fetal increases, allowing for a larger (and easier to genotype) fetal fraction.
This pipeline takes paired end reads generated by an Illumina MiSeq, selects for reads shorter than or equal to the size limit(s) you select, aligns them, and then deduplicates the results.
In-silico Size Selection (ISS) is the process of selecting for shorter reads in a sample. This is performed via Fastp3 4 using the --length_limit flag, followed by the maximum length of nucleotides you are looking for (inclusive.) All subsequent steps will be performed only with the selected reads.
Deduplication is the process of taking multiple copies of a read and creating a single consensus sequence out of them. This balances low amplification and high amplification regions of the genome. This process requires the use of Unique Molecular Idenitifiers (UMIs) to identify the reads. Fastp removes the UMIs and attaches them to the end of the read's ID, and Gencore5 6 uses the UMIs to identify which reads are which.
-
Python 3.10 or higher running on MacOS, a flavor of Linux, or Windows with a Linux Subsystem.
-
The following command line tools installed (all tools were installed via bioconda and use their versioning):
- Fastp
- Burrow-Wheeler Aligner MEM (BWA)
- Genome Analysis ToolKit 4 (GATK4)
- Gencore
-
An appropriate genome reference library, in our case, hg19 or hg38.
-
Fastq.gz files from Paired End samples run on an Illumina MiSeq.
Sample IDs may include a dash (-), but must not contain an underscore (_). If you use an underscore, the sample ID will get truncated. Ex: Sample-ID works, Sample_ID will be truncated to "Sample".
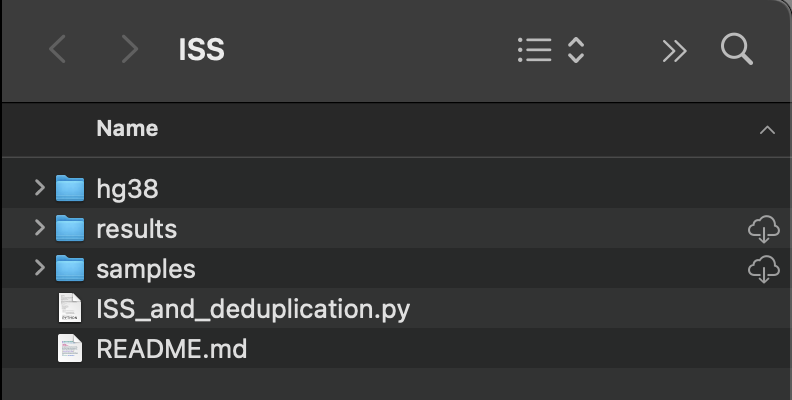
This is the file structure required for this script to run. The gzip'd fastq files from the Illumina MiSeq go in samples, the script's output files are found in the results folder. If you are using a reference genome other than hg38, replace the hg38 folder with the reference genome of your choice. You will also need to update the script accordingly. (More on that below.) The Python script needs to stay in the parent folder.
The fastq.gz files for the deduplication/size selection process can be found here (where Illumina is the base folder where you store your MiSeq results):
Illumina/Name_of_run/Data/Intensities/BaseCalls/sample-name_S#_L001_R1_001.fastq.gz
Illumina/Name_of_run/Data/Intensities/BaseCalls/sample-name_S#_L001_R2_001.fastq.gz
These files should be placed in the samples folder. No prior change or manipulation of the files is necessary. No subfolders are necessary, though files in subfolders will be found and processed as well. This script only works with paired end reads, so you will need R1 and R2 files for each sample.

This script has only been tested using hg19 and hg38, though in theory any reference genome should work as long as it was generated by indexing with the Burrows-Wheeler Alignment tool (BWA), using Samtools' faidx, and then using the Genome Analysis ToolKit 4 (GATK4) CreateSequenceDictionary tool.
The location of the reference genome needs to be specified in the script. In the example in this repository, found in the constants at line 30, the location is specified as follows:
30| REFERENCE_GENOME = Path("./hg38/hg38.fa")Replace the folder and file with your folder location, and the name of the fasta (fa) file within it.
The reference genome folder (hg38) included in this repository is empty except for a .gitignore file, so you'll need to provide your own reference genome.
UMIs are required for deduplication as mentioned in the introduction. The length of the UMI must be specified in the script. The script defaults to 8 nucleotides long, but can be changed if your UMI length is different. This is also set in the constants on line 18.
18| UMI_LENGTH = 8The upper limit of your read lengths from MiSeq data depends on (among other things) the number of cycles you ran the MiSeq with. If you ran the MiSeq for 150 cycles, then you'd have 150 nucleotide reads. However, because we're using 8bp UMIs, and fastp cleaves those off the read, you'd only have 142 nucleotides of data. If you select for 142 nucleotides you will get no size selection, because that is the maximum read length. If you select for 141 nucleotides you will get size selection, the longest reads will not be processed.
\[ number\ of\ MiSeq\ cycles - length\ of\ UMI = Maximum\ read\ length \]
The script will allow you to run as many size selections as you'd like. Line 15 has an array that you enter 1 or more sizes (in nucleotides, inclusive) and will run a cycle of the script for each size for each sample.
15| SIZE_SELECTION_LIST = [135, 140]Using the command line, go to the parent folder with the script in it. The command is simple:
python ISS_and_deduplication.py
Replace python with whatever alias you use to execute Python.
The script will begin looking for samples in the sample folder. A read 1 (r1) file without an accompanying read 2 (r2) file will cause the script to crash.
You do not need to do anything while the script is running.
The script returns the results in the results folder. A single bam file per sample, per size is returned.

Footnotes
-
Lo, Y.M., et al., Presence of fetal DNA in maternal plasma and serum. Lancet, 1997. 350(9076): p. 485-7. ↩
-
Chan, K.C., et al., Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem, 2004. 50(1): p. 88-92. ↩
-
Chen, S. et al., Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics, 2018. 34(17): p. 884–90. https://doi.org/10.1093/bioinformatics/bty560. ↩
-
Chen, S. et al., Gencore: An Efficient Tool to Generate Consensus Reads for Error Suppressing and Duplicate Removing of NGS Data. BMC Bioinformatics, 2019. 20(23): p. 606, https://doi.org/10.1186/s12859-019-3280-9. ↩