We present a computational pipeline to simulate realistic undiagnosed rare disease patients that can be used to evaluate gene prioritization tools. Each simulated patient is represented by sets of candidate disease-causing genes and standardized phenotype terms. We also provide a direct link to download the simulation patients we created using this pipeline.
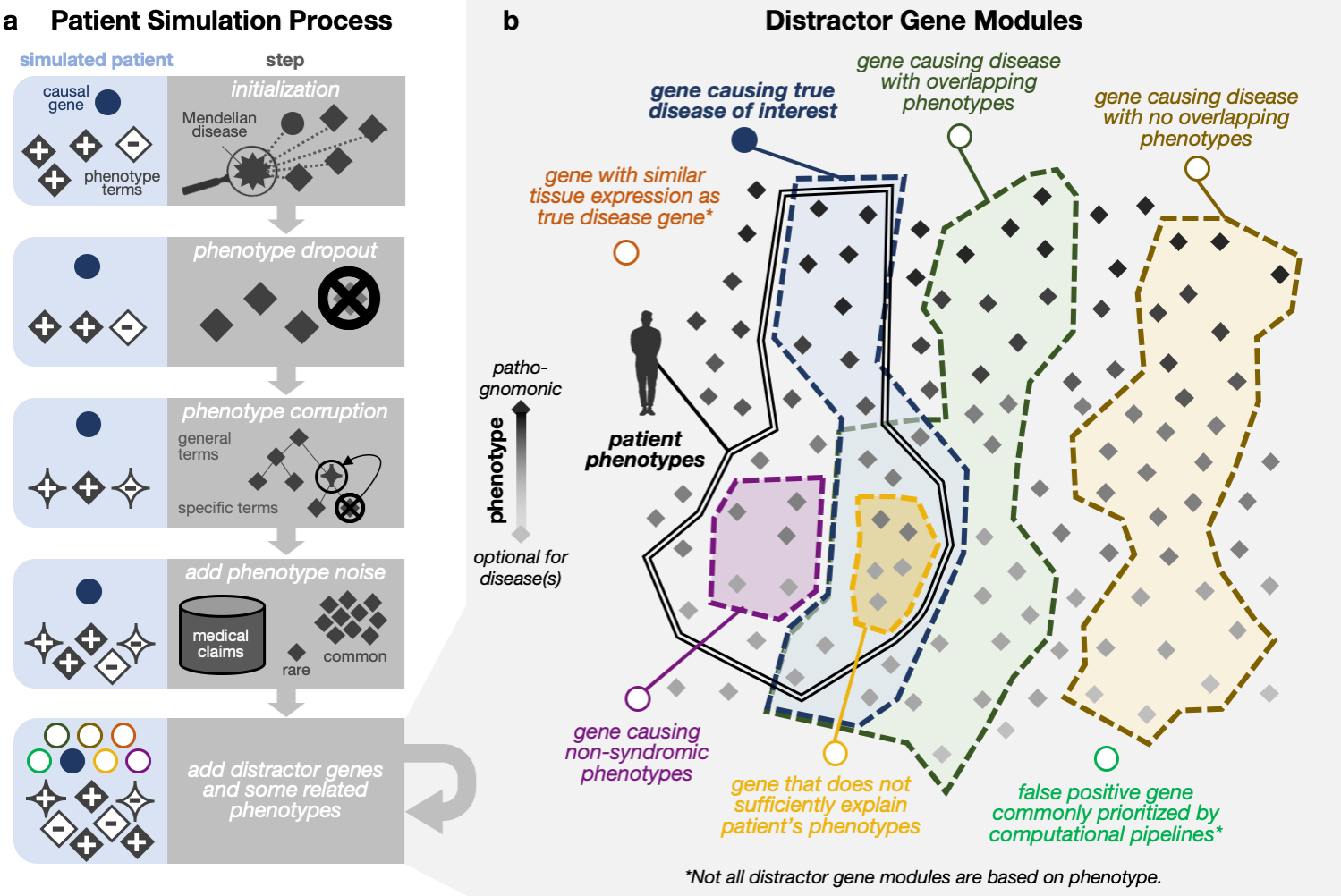
- ✨ Realistic simulation of patient phenotypes and candidate genes: We provide a taxonomy of categories of “distractor” genes that do not cause the patient’s presenting syndrome yet would be considered plausible candidates during the clinical process. We then introduce a simulation framework that jointly samples genes and phenotypes according to these categories to simulate nontrivial and realistic patients.
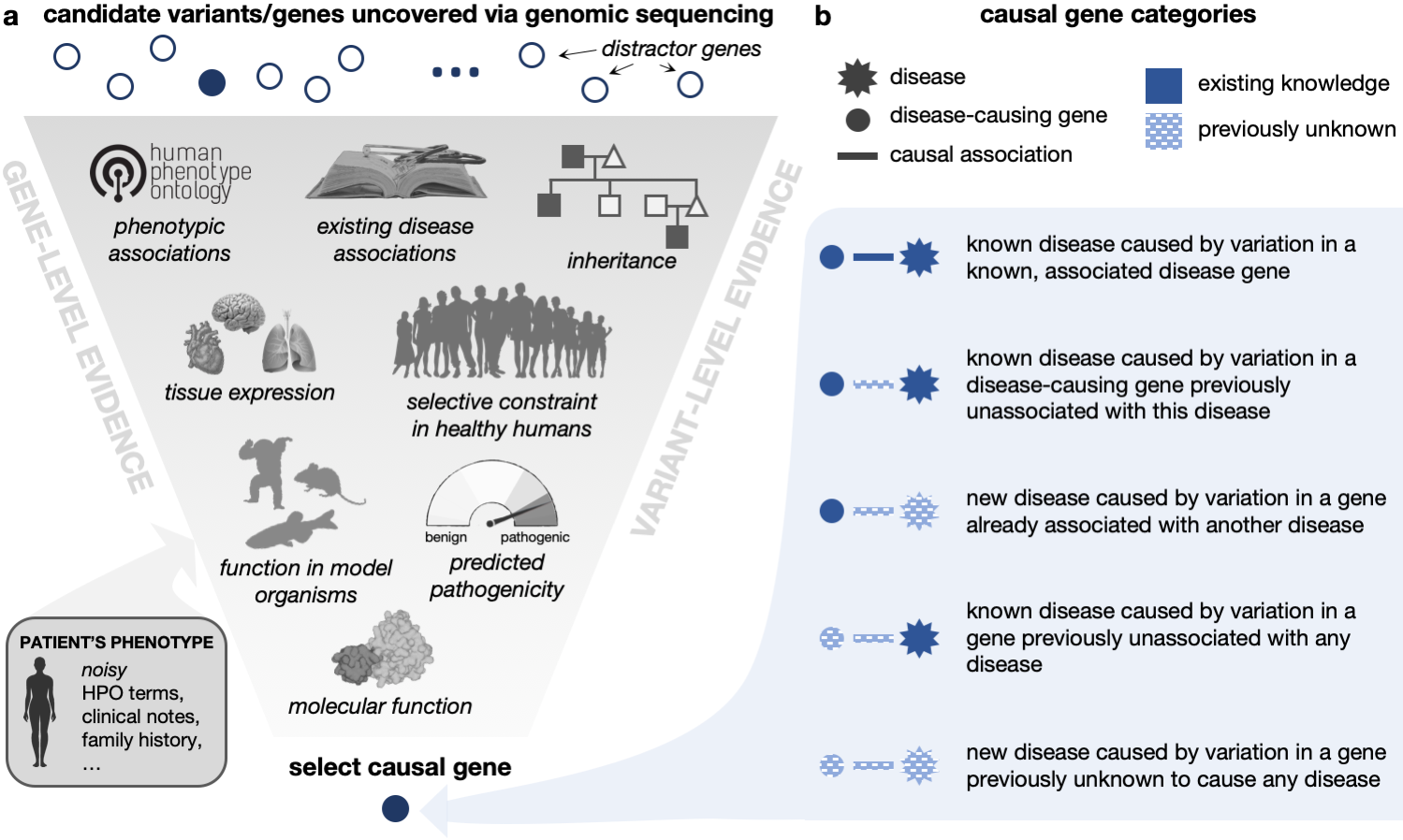
- ✨ Modelling novel genetic conditions: To simulate patients with novel genetic conditions, we curate a knowledge graph (KG) of known gene–disease and gene–phenotype annotations that is time-stamped to 2015. This enables us to define post-2015 medical genetics discoveries as novel with respect to our KG. We manually time-stamp each disease and disease–gene association according to the date of the Pubmed article that reported the discovery and use these time-stamps to annotate each patient according to each of the disease-gene novelty categories below.
- Probing Gene Prioritization Models: We provide our framework and simulated patients as a public resource to enable developers to internally validate and improve their tools by separately evaluating on simulated patients across novelty categories and distractor gene categories.
- Training Machine-Learning based Gene Prioritization Models: We also expect that our dataset of simulated patients that span diverse genetic disorders and reflect realistic clinical processes and imprecision can serve as invaluable training data for machine learning models for rare disease diagnosis.
If you don't want to use the pipeline to simulate your own patients, you can directly download the patients that we simulated to be similar to real patients in the Undiagnosed Diseases Network (UDN). The direct download link is here. Below are statistics comparing the simulated patients to the UDN patients. See the paper for more details.

First, download the github repository.
git clone https://github.com/EmilyAlsentzer/rare-disease-simulation
cd rare-disease-simulation
This codebase leverages Python and many associated packages. To create a conda environment with all of the required packages, ensure that conda is installed and run the following:
conda env create -f environment.yml
conda activate rare-dx-env
This process should take no more than a few minutes. Note that this has been tested on linux software.
After the conda environment is created and activated, install the github repo with the following:
pip install -e .
Before you run any code in the repository, you will need to (1) download the required data and (2) specify the path to the project directory.
The data is hosted on Harvard Dataverse with the persistent identifier: https://doi.org/10.7910/DVN/ANFOR3. To maintain the directory structure while downloading the files, make sure to select all files and download in the original format. The entire dataset is approximately 3GB when zipped. Note that if you only wish to download the already generated simulated patients, you can directly download simulated_patients_formatted.jsonl.
Go to config.py and set the project directory (PROJECT_ROOT) to be the path to the data folder downloaded in the previous step.
To run the simulation pipeline with default parameters, run the following code. This will run the simulation pipeline and output the simulation patients to a jsonl file named simulated_patients.jsonl by default in the directory specified by config.SIMULATED_DATA_PATH. Generation of the simulated cohort with default parameters takes ~1 hr, but you can modify the # of patients produced in the config.py file.
python simulation_pipeline/simulate_patients.py
The parameters of the simulation pipeline and their associated descriptions can be found in the config.py file. Modify these parameters to change the number of patients produced, the frequency of sampling each distractor gene module, etc.
The simulated patients are saved as a json lines file, i.e. each line in the file is a json containing the information for a single patient. Below is a schema for the json and an example json for a single simulated patient.
{
"id": int,
"disease_id": str, // orphanet ID
"true_genes": list(str), // list of ensembl ids for the patient's causal gene(s)
"age": str, // age category
"positive_phenotypes": {str : list(str)} // dict mapping phenotype to list of simulation modules that added the phenotype
"distractor_genes": {str : list(str)} // dict mapping ensembl id to list of simulation modules that added the distractor gene
"n_distractor_genes": int, //count of # distractor genes
"dropout_phenotypes": {str : list(str)} // dict with two entries for positive &. negative phenotypes that keep track of which phenotypes were removed during the dropout stage. This is important for the ablation analysis to be able to retroactively add back in the dropped phenotypes
"corruption_phenotypes": {str : list(str)} // dict with two entries for positive &negative phenotypes that keep track of which phenotypes were corrupted during the corruption stage. This is important for the ablation analysis to be able to retroactively "un-corrupt" the phenotypes
}
{
"id": 9,
"disease_id": "966",
"true_genes": ["ENSG00000069431"],
"age": "Onset_Adult",
"positive_phenotypes": {"HP:0000221": ["init_phenotypes"], "HP:0000232": ["init_phenotypes"], "HP:0001155": ["init_phenotypes"], "HP:0005692": ["init_phenotypes"], "HP:0012471": ["init_phenotypes"], "HP:0100540": ["init_phenotypes"], "HP:0001999": ["phenotype_corruption"], "HP:0001249": ["phenotype_distractor.1", "phenotype_distractor.3", "insufficient_explanatory.11"], "HP:0010285": ["phenotype_distractor.12"], "HP:0000924": ["non_syndromic_phenotype.13"], "HP:0004459": ["noisy_phenotype"]}, "negative_phenotypes": {"HP:0002615": ["phenotype_distractor.1"], "HP:0012734": ["phenotype_distractor.1"], "HP:0007440": ["phenotype_distractor.1"], "HP:0011043": ["phenotype_distractor.1"], "HP:0000470": ["phenotype_distractor.3"], "HP:0010978": ["phenotype_distractor.3"], "HP:0000902": ["phenotype_distractor.3"], "HP:0000347": ["phenotype_distractor.12"], "HP:0000272": ["phenotype_distractor.12"], "HP:0004209": ["phenotype_distractor.12"], "HP:0010492": ["noisy_phenotype"]},
"distractor_genes": {"ENSG00000165125": ["pathogenic_pheno_irrel.0"], "ENSG00000184470": ["phenotype_distractor.1"], "ENSG00000108821": ["pathogenic_pheno_irrel.2"], "ENSG00000179111": ["phenotype_distractor.3"], "ENSG00000157766": ["pathogenic_pheno_irrel.4"], "ENSG00000104899": ["pathogenic_pheno_irrel.5"], "ENSG00000157119": ["pathogenic_pheno_irrel.6"], "ENSG00000168646": ["pathogenic_pheno_irrel.7"], "ENSG00000142655": ["pathogenic_pheno_irrel.8"], "ENSG00000143156": ["tissue_distractor.9"], "ENSG00000131828": ["pathogenic_pheno_irrel.10"], "ENSG00000196277": ["insufficient_explanatory.11"], "ENSG00000088451": ["phenotype_distractor.12"], "ENSG00000157557": ["non_syndromic_phenotype.13"]},
"n_distractor_genes": 14,
"dropout_phenotypes": {"positive_phenotypes": ["HP:0000414"], "negative_phenotypes": ["HP:0000212", "HP:0000581", "HP:0001249", "HP:0002230", "HP:0010285"]},
"corruption_phenotypes": {"positive_phenotypes": ["HP:0000280"]}
}
To annotate the simulated patients from the prior step with their disease-gene novelty category, run:
python simulation_pipeline/label_patients_with_novelty_categories.py --input simulated_patients.jsonl
This will output another jsonl file that ends in _formatted.jsonl in which each json has the same entries as above, but also includes additional labels that denote the patient's disease-gene novelty category (e.g. known_gene_disease) and whether the patient's gene is found in the knowledge graph used to evaluate the gene prioritization methods.
The simulated patients can be run on any gene prioritization model that accepts sets of phenotypes and (optionally) sets of candidate genes. We provide an example of how to run Phrank on the simulated patients below.
To run both versions of the Phrank algorithm:
sh gene_prioritization_algorithms/run_phrank.sh
If you would like to evaluate the simulated patients on algorithms that require VCFs as input, you can leverage the script create_vcfs.py in the create_vcfs folder to sample a variant for each candidate gene and construct a VCF.
The evaluation script assumes that the gene prioritization results are saved as a pickle file containing a dict mapping from patient id to a list of tuples where each tuple contains (gene_score, candidate_gene_name).
Run the evaluation script with the following, replacing PATH_TO_RESULTS_PKL with the path to the pickle file outputted from the gene prioritization algorithm in the prior step. This script will print the performance to the terminal and save a figure with top-k performance across novelty categories.
python evaluation/evaluate.py --patients simulated_patients_formatted.jsonl --results PATH_TO_RESULTS_PKL
5️⃣ Create, run, and evaluate on simulated patients created with portions of the simulation pipeline removed
Run the following to reproduce our simulation pipeline ablation experiment, i.e. to create many versions of the simulated patients in which different components of the simulation pipeline are removed.
python simulation_pipeline/simulate_patients.py --sim_many_genes --equal_probs
python simulation_pipeline/simulate_patients.py --random_genes
sh simulation_pipeline/perform_ablations.sh
This will first create two cohorts of simulated patients: (1) patients with many candidate genes and (2) patients with random candidate genes. The bash script generate different versions of the simulated patients by ablating the phenotype and gene modules in the pipeline. In the gene ablations, the candidate genes of the patients created in step 1 will be filtered to include only the genes from the specified gene modules and replaced with randomly sampled genes from the patients generated in step 2.
After the ablated simulation patients are created, you can run and evaluate the gene prioritization models on each dataset as in usage 3 & 4 above. Running gene prioritization algorithms on the gene module ablations could prove useful for determining whether there are specific categories of distractor genes in which the model performs most poorly.
To run Phrank on the the ablated patients, modify the PATIENT_PATH in gene_prioritization_algorithms/run_phrank_ablations.sh and then run the script. To evaluate, you can either pass in a single ablated patient jsonl and results path to evaluate.py as in usage 4. Alternatively, to evaluate and plot all ablations at once, run:
python evaluate_ablations.py \
--results_suffix '_phrank_rankgenes_directly=False.pkl' # suffix added on to each ablation run for the gene prioritization algorthm (here, Phrank Patient-Disease)
--results_path /PATH/TO/ABLATION/RESULTS # replace with a path to all of the ablation results - e.g. config.SIMULATED_DATA_PATH / 'gene_prioritization_results' / 'phrank_ablation'
- In order to perform the time-stamped evaluation, we manually time-stamped each disease and disease–gene association in Orphanet according to the date of the Pubmed article that reported the discovery. We release these annotations publicly to the community in the data download in the Harvard Dataverse.
- We also provide our preprocessing code that we used to (1) preprocess all orphanet data and (2) convert all genes to Ensembl IDs and update all HPO terms to the 2019 Human Phenotype Ontology. These preprocessing scripts can be found in the
process_simulation_resourcesfolder, but we also provide the already processed data in the data download.
This project is covered under the MIT License.
For questions, please leave a Github issue or contact Emily Alsentzer at ealsentzer@bwh.harvard.edu.