![]()
A Landmark-based Approach for Generating Multi-cellular Sample Embeddings from Single-cell Data
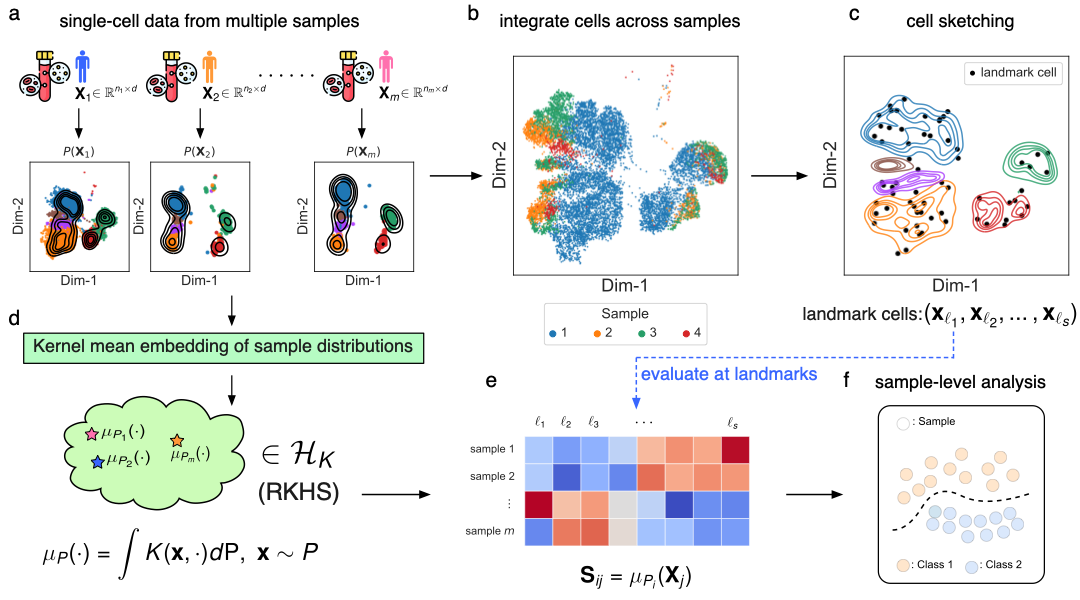
scLKME is a computational approach designed for generating sample embeddings in multi-sample single-cell data. scLKME models samples as cell distributions, and leverages lanbmark-based kernel mean embedding to generate a sample-by-landmark kernel matrix as sample embeddings. The workflow of scLKME is as follows:
scLKME requires python version >= 3.8, < 3.12.
scLKME including its dependencies can be installed via PyPI by running:
pip install sclkmeAll the datasets used in this study are shared via .h5ad format. The datasets are available at:
scLKME's apis are designed to be compatible with the scanpy's ecosystem. To use scLKME, here is a basic example:
import sclkme
import scanpy as sc
n_sketch= 128
sample_id = "sample_id"
adata = sc.read_h5ad("path_to_h5ad_data")
sclkme.tl.sketch(adata, n_sketch=n_sketch)
X_anchor = adata[adata.obs['sketch']].X.copy()
sclkme.tl.kernel_mean_embedding(adata, partition_key="sample_id", X_anchor=X_anchor)
# sample_embedding
adata.uns['kme'][f'{sample_id}_kme']For more details, examples and tutorials, check our document.
| Tutorials | Colab |
|---|---|
| Cell Sketching | |
| Landmark-based multi-sample single-cell data analysis | |
@article{yi2023sclkme,
title={scLKME: A Landmark-based Approach for Generating Multi-cellular Sample Embeddings from Single-cell Data},
author={Yi, Haidong and Stanley, Natalie},
journal={bioRxiv},
pages={2023--11},
year={2023},
publisher={Cold Spring Harbor Laboratory}
}