This repository contains a script which performs simulations of Coulomb clusters confined in a harmonic potential.
These simulations lead to simple, "polyhedral" structures if the system is cooled to low enough temperatures. The program aims to help visualize these structures by finding the potential energy minimum of the system and its geometry by using the Monte Carlo method.
The code is VERY unoptimized as of now, but it will be improved in the future. The potential energy calculations are performed using the GPU by means of the numba python module.
These instructions will get you a copy of the project up and running on your local machine for development and testing purposes.
The script works with python versions > 3.6, mainly because it uses f-strings for returning some results.
Additionally, the script uses several python modules. These modules are listed on the "requirements.txt" file, and can be installed by running:
python3 -m pip install -r requirements.txt
while the file is located in the cwd.
Additionally, for the GPU calculations to work, installing the CUDA toolkit is required. For example, in a Ubuntu installation the toolkit can be installed with the following command:
apt install nvidia-cuda-toolkit
check the official the numba installation guide for additional information.
Running the script is simple. After cloning the repository, the script run_simulation.py inside the "CoulombChargesTrap3D" folder should be executed with:
python3 run_simulation.py
The program will run the simulation and save the results in a new folder named 'iontrap_TIMESTAMP'.
As of now, the script run_simulation.py must be changed manually to modify any parameters of the system.
An argument parser may be included in the future, but for now, the values of the dictionary "ar_dict" inside the run_simulation script must be changed manually.
Here is a quick overview of the parameters:
- N_list: List of different number of particles. This will make the script run various simulations for the given N. Each N must be an integer.
- L: Lattice size (float)
- q: Ionic charge (float)
- m: Mass (float)
- alpha: Symmetry of the external harmonic potential. Alpha = 1 for spherical symmetry and alpha != 1 for cylindrical symmetry. (float)
- omega: Natural frequency (float)
- T: Initial Temperature (float)
- T_f: Target temperature for the annealing process. (float, this needs to be approximately zero to be able to ignore the kinetic energy contribution)
- confs: Number of initial configurations to prepare for the MC calculations (int)
- plot: Whether to prepare plots for the results or not (bool)
The file mc_utils.py contains the main functions necessary for the simulation and it is intended to be used as a module file for the run_simulation.py script.
Here are some of the obtained geometries for nMCS = 10^7 and for different number of ions (N):





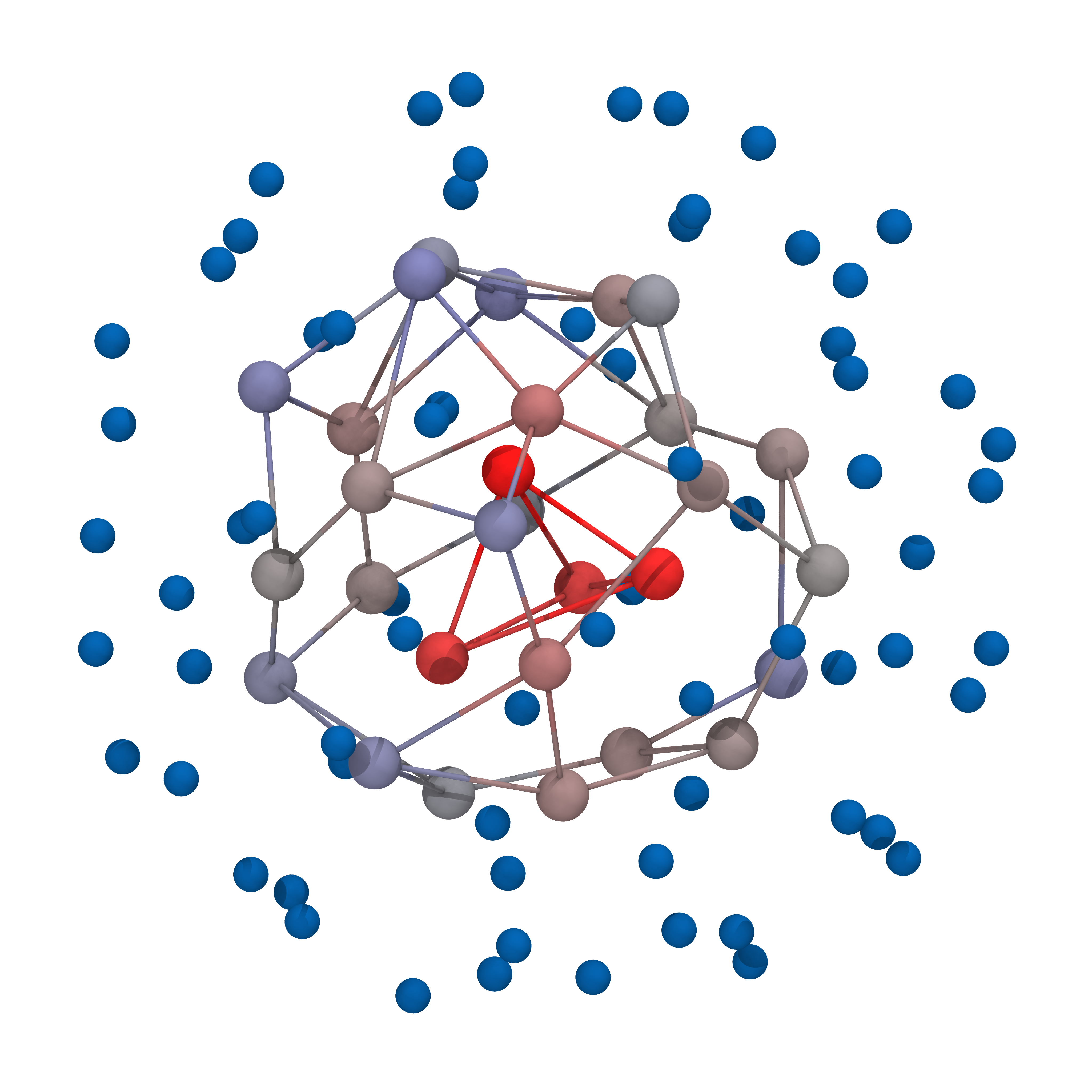
-
Lozovik, Yu. E., & Mandelshtam, V. A. (1990). Coulomb clusters in a trap. En Physics Letters A (Vol. 145, Issue 5, pp. 269-271). Elsevier BV.
-
Rafac, R., Schiffer, J. P., Hangst, J. S., Dubin, D. H., & Wales, D. J. (1991). Stable configurations of confined cold ionic systems. In Proceedings of the National Academy of Sciences (Vol. 88, Issue 2, pp. 483–486).